In order to organize your data according to GeneViTo
format you must follow these instructions :
File Formats
1. Essential files
In order to use the GeneViTo
GUI you must provide at least
the following files :
GenBank files
*.ptt = Protein Table
*.ffn = FASTA nucleotide coding regions
file
*.fna = FASTA Nucleic Acid file
These files are available through the GenBank FTP
site : ftp://ftp.ncbi.nih.gov/genbank/genomes/
e.g.
In order to acquire the above mentioned files for
Chlamydia trachomatis you have to visit ftp://ftp.ncbi.nih.gov/genbank/genomes/Bacteria/Chlamydia_trachomatis/
, where all these files are stored.
Proteome file
The proteome file must follow the
SWISS-PROT format. This file can be acquired for example from the SWISS-PROT
database by downloading
through the SRS system http://srs.ebi.ac.uk/
the entire proteome or from the Proteome Analysis Server at EBI http://www.ebi.ac.uk/proteome/index.html?http://www.ebi.ac.uk/proteome/ProteomeSource.html.
e.g.
ID
6PGD_CHLTR STANDARD; PRT; 480 AA.
AC O84066;
DT 30-MAY-2000 (Rel. 39, Created)
DT 30-MAY-2000 (Rel. 39, Last sequence update)
DT 30-MAY-2000 (Rel. 39, Last annotation
update)
DE 6-PHOSPHOGLUCONATE DEHYDROGENASE,
DECARBOXYLATING (EC 1.1.1.44).
These files are not essential for the GUI to
work. They supply additional information on genomic/proteomic elements. These
files are :
Structural
RNAs (GenBank file)
This file is available through http://www.ncbi.nlm.nih.gov/genomes/static/eub_g.html.
By selecting the genome of choice you have to :
e.g. for Clamydia trachomatis , click on
the second column (NC_000117).
In the new page that appears you must select from the feature table
the Structural
RNAs option. Doing so, a new page will open and from the
the report below in format. menu you have to select FASTA nucleotide and
save it in a plain text file.
Clusters of
Orthologous Groups (COGs file)
This file is available through http://www.ncbi.nlm.nih.gov/genomes/static/eub_g.html.
By selecting the genome of choice you have to :
e.g. for Clamydia trachomatis , click on
the second column (NC_000117).
In the new page that appears you must select from the top right column
BLAST protein homologs: COGs (Clusters
of Orthologous Groups) 3D
Structure (Sequences with known structure) TaxMap (Sequences
grouped by superkingdom) TaxPlot (3-way
genome comparison) CDD(Conserved
Domain Database)
the COGs (Clusters
of Orthologous Groups) option, that will open a new page. On this new page the
COGs functional classes are displayed for the aforementioned organism.
You have to click on the second column COGs
and a new page appears. This page must be saved in a .txt file
(selecting from the browser option Save As). This must be done for all the
categories and the relative files must have the following filenames:
Amino acid transport and metabolism.txt
Carbohydrate transport and metabolism.txt
Cell cycle control, mitosis and meiosis.txt
Cell motility.txt
Cell wallmembrane biogenesis.txt
Chromatin structure and dynamics.txt
Coenzyme transport and metabolism.txt
Cytoskeleton.txt
Defense mechanisms.txt
Energy production and conversion.txt
Extracellular structures.txt
Function unknown.txt
General function prediction only.txt
Inorganic ion transport and metabolism.txt
Intracellular trafficking and secretion.txt
Lipid transport and metabolism.txt
Nuclear structure.txt
Nucleotide transport and metabolism.txt
Posttranslational modification, protein turnover, chaperones.txt
RNA processing and modification.txt
Replication, recombination and repair.txt
Secondary metabolites biosynthesis, transport and catabolism.txt
Signal transduction mechanisms.txt
Transcription.txt
Translation.txt
not in COGs.txt
These files must be put inside a directory.
Provide a name of your choice for the directory.
Translation Table (GenBank file)
This file is available through http://www.ncbi.nlm.nih.gov/genomes/static/eub_g.html.
By selecting the genome of choice you have to :
e.g. for Clamydia trachomatis , click on
the second column (NC_000117).
In the new page that appears you must select under the circular map
TTT F Phe TCT S Ser TAT Y Tyr TGT C Cys
TTC F Phe TCC S Ser TAC Y Tyr TGC C Cys
TTA L Leu TCA S Ser TAA * Ter TGA * Ter
TTG L Leu i TCG S Ser TAG * Ter TGG W Trp
CTT L Leu CCT P Pro CAT H His CGT R Arg
CTC L Leu CCC P Pro CAC H His CGC R Arg
CTA L Leu CCA P Pro CAA Q Gln CGA R Arg
CTG L Leu i CCG P Pro CAG Q Gln CGG R Arg
ATT I Ile i ACT T Thr AAT N Asn AGT S Ser
ATC I Ile i ACC T Thr AAC N Asn AGC S Ser
ATA I Ile i ACA T Thr AAA K Lys AGA R Arg
ATG M Met i ACG T Thr AAG K Lys AGG R Arg
GTT V Val GCT A Ala GAT D Asp GGT G Gly
GTC V Val GCC A Ala GAC D Asp GGC G Gly
GTA V Val GCA A Ala GAA E Glu GGA G Gly
GTG V Val i GCG A Ala GAG E Glu GGG G Gly
Coding GC 41.53% 1st letter GC 51.56% 2nd letter GC 39.20% 3rd letter GC
33.85%
The "red"
highlighted region above, must be saved (Copy & Paste) in a text
file with a name of your choice .
Webcutter
(Restriction Enzymes file)
At the Webcutter
website http://www.firstmarket.com/cutter/cut2.html,
you must have selected the following options before running Webcutter :
Here is a sample of
Webcutter output:
AatI 7 9333 13482 19521 42397 48711 agg/cct More info
56867 57843
AatII 3 1929 49019 80792 gacgt/c More info
Acc113I 12 6298 11712 17493 24282 31362 agt/act More info
32362 38424 45260 76393 76399
79306 80868
Acc16I 17 3122 3333 17333 19198 20267 tgc/gca More info
27062 28425 29882 40655 46328
59270 61994 63929 66818 68610
71292 74072
For an entire genome
you must provide a file with all the desired restriction enzymes in the above
("yellow region") format. Bear in mind, that Webcutter, accepts a
limited number of sequences for a batch run. Thus, when you
"run" a whole genome in sets of sequences, the relative cut position
(in bp) of the input sequences is reset each time. For example, providing a set
that corresponds to 1000-2000bp of a genome, the output file will start the
numbering of restriction enzymes cut sites from position 1bp. The GeneViTo
file format requires that you have a single file,
with the above format, and the cut-positions should correspond to their
actual position on the genome.
PRED-CLASS
results file
The PRED-CLASS
algorithm is available through http://biophysics.biol.uoa.gr/PRED-CLASS/
.
Now, you can :
Run PRED-CLASS
on a sequence
Browse
detailed results obtained with the algorithm on several test sets
View
lists of data used to train and evaluate PRED-CLASS
Go to the Biophysics Lab Homepage
You have to
select Run PRED-CLASS on a sequence, in order to run PRED-CLASS. In the new page
that appears you must insert the protein in the provided area. This option
provides one by one protein "run". For an entire proteome, upon
request to the PRED-CLASS authors, batch run might be available. However, a list
of PRED-CLASS results for 21 prokaryotic and 6 archaean entire proteomes
is available through http://biophysics.biol.uoa.gr/PRED-CLASS/Results/FULL/
(a README file provides further info).
The GeneViTo
file format for PRED-CLASS results is that of the files in
http://biophysics.biol.uoa.gr/PRED-CLASS/Results/FULL/.
You must provide a plain .txt file with this format.
PRED-TMR2
& orienTM results file
The PRED-TMR2 algorithm is available through http://biophysics.biol.uoa.gr/PRED-TMR2/
.
Now, you can :
Run PRED-TMR2
on a sequence
Browse
the results obtained with the algorithm
Go to the Biophysics Lab Homepage
You have to select Run PRED-TMR2 on a sequence,
in order to run PRED-TMR2. In the new page that appears you must insert the
protein in the provided area. This option provides one by one protein
"run". For an entire proteome, upon request to the PRED-TMR2
authors, batch run might be available. However, a list of PRED-TMR2 results
for 7 entire proteomes is available through http://biophysics.biol.uoa.gr/PRED-TMR2/Results/index.html.
The orienTM algorithm is available through http://biophysics.biol.uoa.gr/orienTM/.
You have to select Execute
orienTM on a sequence in order to run orienTM on a
protein sequence with already defined transmembrane segments. You can also run
PRED-TMR2 and orienTM successively.
The GeneViTo
file format for PREDTMR2 & orienTM is as follows (in
XML format):
<SEQ>
<NAME>AROB_CHLTR </NAME>
<RESULTS>
<TM>
<FROM>93</FROM>
<TO>112</TO>
<ORIEN>OUTWARDS</ORIEN>
</TM>
</RESULTS>
</SEQ>
Following the above mentioned XML format( you must comply to
the above format strictly i.e. the respective tags must be on the same
lines as in the example for a protein - there can't be for example
<FROM> and </FROM> tags in different lines), you can insert
prediction results from various algorithms e.g. in the <TM> tag, you can
insert predicted transmembrane segments in the <FROM> </FROM>
and <TO> </TO> tag groups.
A <SEQ> tag, can contain one <NAME> </NAME>
tag group, one <RESULTS> </RESULTS> tag group. Inside the RESULTS
tag group there can be several <TM> </TM> tag groups. Inside the TM
group, there can be several <FROM> </FROM> tags followed by
<TO> </TO> tags, followed by <ORIEN> </ORIEN> tags. In
case you don't have one of the elements you fill in a "-" among
the tags.
SIGNALP results file
The SIGNALP algorithm is available through http://www.cbs.dtu.dk/services/SignalP/.
You have to run either one by one or batch, all the proteome and provide a
single file with the results following the typical SIGNALP format:
>DUT_CHLTR
SignalP-NN result:
>DUT_CHLTR length = 70
# Measure Position Value Cutoff signal peptide?
max. C 18 0.367 0.50 NO
max. Y 18 0.113 0.32 NO
max. S 4 0.433 0.90 NO
mean S 1-17 0.184 0.44 NO
SignalP-HMM result:
>DUT_CHLTR
Prediction: Non-secretory protein
Signal peptide probability: 0.015
Max cleavage site probability: 0.009 between pos. 23 and 24
>EFG_CHLTR
SignalP-NN result:
>EFG_CHLTR length = 70
# Measure Position Value Cutoff signal peptide?
max. C 41 0.105 0.50 NO
max. Y 41 0.083 0.32 NO
max. S 70 0.286 0.90 NO
mean S 1-40 0.084 0.44 NO
SignalP-HMM result:
>EFG_CHLTR
Prediction: Non-secretory protein
Signal peptide probability: 0.000
Max cleavage site probability: 0.000 between pos. -1 and 0
Inserting the above files in GeneViTo
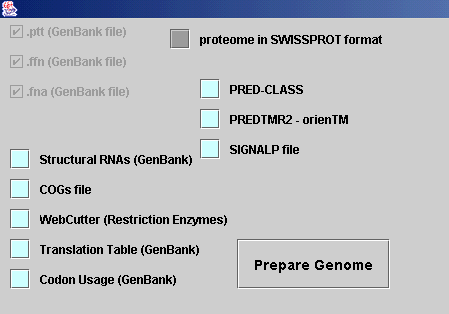
The input of all file is done through this
interface:
1. Selecting Essential files
From the menu File you choose the option Prepare
Genome and the above interface appears.
If you click Prepare Genome without
selecting no other feature, you will be prompted to insert through File Choosers
the Essential Files mentioned at the beginning of this document. These
files (.ptt, .ffn, .fna, proteome in SWISSPROT files) are obligatory
for the genome to be prepared. In this procedure you will be prompted to provide
a name for the project you are preparing, along with the Destination of
the Directory that will be created.
This procedure is standard and will take
place each time you prepare a genome, regardless the Additional input files
choices you have made.
2. Selecting Additional files
According to the data you have at hand (which
additional files did you gather following the procedure mentioned above?), you
have to select the relative cyan toggle buttons, and then enter the
corresponding files, through File Choosers. Afterwards, you have to push the Prepare
Genome button, enter the Essential files and wait for your genome to be
prepared.
In this last picture you can see the procedure of
locating a file through a File Chooser.