


|
3d Structure Fit |
Palaios, G.A. and Hamodrakas, S.J. |
|
Index | |
2.Copyright notice | |
4. Description of the Output |
The input page, which allows a user to specify the data needed for the program, consists of two pages:
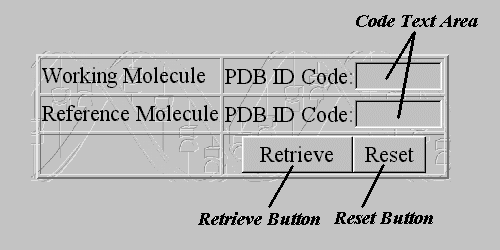
The Retrieve Form actually consists of two text fields. In the first one the user must enter the Working Molecule's PDB ID Code whereas in the second the Reference Molecule's PDB ID Code. If the user leaves one of the text fields blank the program produces an Error Message and aborts the procedure. If one of the PDB ID Codes does not exist the program returns to the main Input Page.
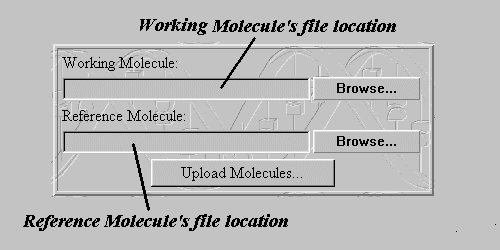
The 'Browse...' buttons of the Upload Form give the user the ability to choose the exact location of the two PDB Files that contain the Working and the Reference Molecules atomic coordinates. By clicking the 'Upload Molecules...' button the user activates the script that gets the contents of this files and proceeds to the next step.
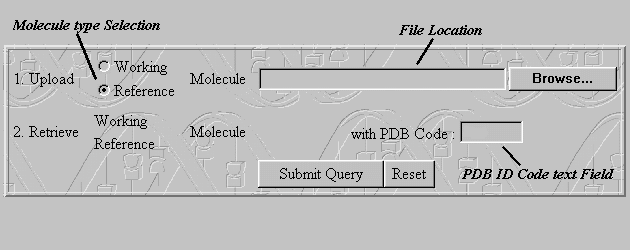
This form is made for users that have one of the files stored(files containing the Working Molecule or Reference Molecule) localy on their hard disk. In that case first select the type of the file (Working Molecule or Reference Molecule) that you wish to upload. Use the 'Browse...' Button to enter the exact location of the file stored on your hard disk and then enter the PDB ID Code of the file you wish to download from the Protein Databank. Click on the 'Submit Query' Button to activate the programs
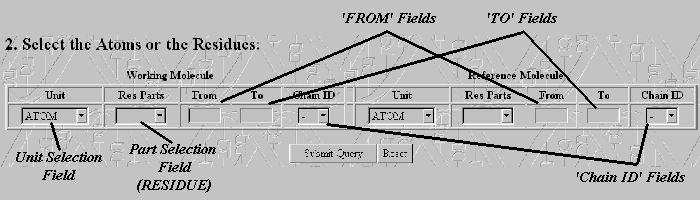
The LSQR's 'Generate' Module first checks the contents of the Input Files and prints in a table information about the stored structures such as their 'HEADER' and 'COMPND' Lines and the total number of atoms that appear in the file. If no 'ATOM' lines are detected the program produces an Error message and aborts. If the Input Files are valid the program produces the Command Panel. The Command Panel's layout is shown below. The user can select to match either 'ATOMS' or 'RESIDUES' for both Working and Reference Molecules by choosing the appropriate option from the Unit Select Field. If the 'RESIDUE' option is selected the user is capable to choose the part of the residues that are going to be used by the program. The user must choose between the options of the table below:
| Option | |
| MAIN | Select to use only the Main Chain |
| SIDE | Select to use only the Side Chain |
| ALL | Select to use All the Atoms of the Residues |
| CA | Select to use only the Ca Atoms |
If the structures consist of more than one chains the user must enter in the Chain ID Field
the ID of the Chain to be used. In the 'FROM' and 'TO' text fields
the user must enter the number of the Start Atom or Residue and the number
of the End Atom or Residue for the Working and the Reference Molecules. Value 'TO' must always be more
than Value 'FROM' and the length of the segment selected for the Working Molecule must be equal to the length
of the segment selected in the Reference Molecule. If those conditions are not satisfied the program produces
Error messages, removes the Input Files and stops the execution.
Error messages are displayed in a new HTML Error Page.
Explanation of the error messages:
 |
 |
 |
 |